CHEM0028: COMPUTATIONAL CHEMISTRY
Extended Assessment
24 January 2022
SECTION A
1. Polyimides are polymers which typically have very high melting temperatures
compared to most other polymers (> 200 °C). A general overview can be found in Wikipedia for background information (https://en.wikipedia.org/wiki/Polyimide). These materials typically form semi-crystalline structures – you should refer to this article for an overview of structure of polymers: https://en.wikipedia.org/wiki/Crystallization_of_polymers.
No further reading or knowledge of polymers is required here beyond A-level chemistry.
Consider the series of related polyimides (A to D) which differ only in the structure of the link between the imide groups. Experimentally the value of n is of the order of 2000.
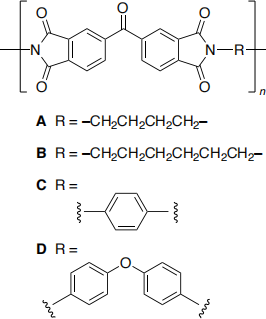
Outline a computational study based on molecular mechanics forcefield methods to investigate which of the materials (A to D) is most likely to form the higher melting point material and which forms the most crystalline (regularly ordered arrangement of polymer chains) material.
Your study should consider:
. How to construct models of the polymer.
. How to determine the preferred conformational structures of each polymer.
. How might you treat different crystalline regions.
. How you might simulate the melting of a model of each polymer structure you consider and how you would determine the relative melting temperature of these
models from your simulations.
. Whether the study of a single repeat unit (i.e. the molecule where n = 1 terminated with hydrogen) would provide sufficient information to deduce the order of melting temperatures.
. The interactions that the forcefield must be able to describe accurately for your study (although explicit selection of a forcefield is not required).
No calculations are expected. You may use the chemical drawings which are included in the appendix and here.
Note that you should consider for the purpose of this question that to simulate a “realistic (experimental)” single polymer chain (molecule) or assembly of chains is not feasible. The longest single chain that can be simulated is n = 10 and an assembly of at most 20 such chains can be constructed and studied. The ends of a single polymer chain can be considered a hydrogen atom.
Note also that polymers do not form perfect crystals. Rather they have regions where there is regular packing of polymer chains. Moreover, a chain may form a regular repeating arrangement with itself (see scheme A) as well as between different chains.
Scheme A:
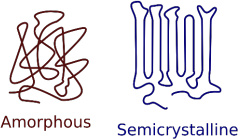
https://commons.wikimedia.org/w/index.php?curid=10491297
[30 marks]
SECTION B
2. (a) Briefly describe the Hartree Product and Slater Determinant approaches to writing the wavefunction of a multielectron system. Which approach is usually used and why? [6 marks]
(b) Compare and contrast the main principles/assumptions of Hartree-Fock theory and Density Functional Theory (DFT). By comparing the Hartree-Fockeigenvalue equation and the Kohn-Sham orbital eigenvalue equation, or otherwise, consider the extent to which these methods account for
. Electron kinetic energy
. Electron correlation
. The Coulomb interaction
. The exchange interaction
. Electron-nuclear attraction
Note: for this part of the question you are not expected to comment on excited state calculation. [14 marks]
(c) Hydrogen Fluoride (HF) is believed to polymerize in the gas phase. Assuming a computational framework of either Restricted Hartree-Fock theory or Density Functional Theory, suggest what calculations using which basis sets should be used to compute the binding energy of a HF dimer as shown below:
H F - - - --H F
[5 marks]
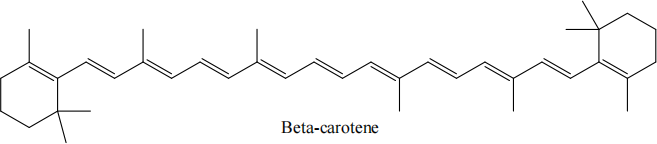
(d) The lowest excited singlet (!! ) state of polyenes, such as beta carotene shown below, are believed to have substantial contributions from double excitations, i.e. the lowest excited state is formed by exciting both electrons in the HOMO to the LUMO. Suggest one electronic structure method which could be used to compute the !! excitation energy in beta carotene. Briefly describe the method, explaining its suitability for this system. Suggest any advantages this method has and any challenges the calculation may encounter. [5 marks]
SECTION C
3. The figure below shows the structure of aniline and the average number of aniline molecules adsorbed onto a hexagonal ice surface as a function of chemical potential, calculated using a Monte Carlo simulation.
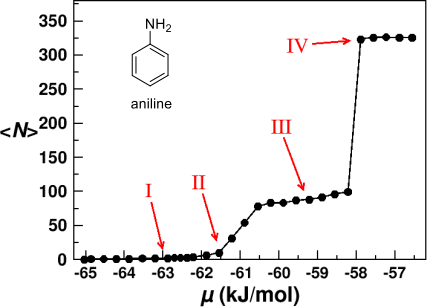
Adsorption isotherm showing the average number of aniline molecules per unit cell adsorbing onto a hexagonal ice surface as a function of the chemical potential µ at 200 K
(from Fu et al., J. Mol. Liq. 290 (2019) 111221).
The adsorption isotherm in the figure above is an example of multi-layer adsorption of small molecules. Describe a protocol for simulating adsorption of small molecules on a surface using Metropolis Monte Carlo (MMC) and explain why MMC is a suitable computational technique for simulating adsorption isotherms and examining the molecular processes involved. Your answer should include:
(a) A list of the steps involved in performing a Metropolis Monte Carlo (MMC) simulation of a molecular system, both in general and more specifically for the aniline adsorption on hexagonal ice system described above. [10 marks]
(b) A description of the different thermodynamic ensembles MMC can simulate and an explanation of the most appropriate MMC ensemble to calculate adsorption isotherms. [6 marks]
(c) An explanation of why MMC is more suitable for calculating adsorption
isotherms than molecular dynamics methods, emphasising the differences in the two techniques. [6 marks]
(d) A description of the nature of the assemblies of molecular structure that are formed on adsorption in the example above, as the chemical potential is varied. There should be reference to the four distinct regions I to IV indicated in the figure. State any limitations you might encounter in achieving the behaviour observed for the high-density state IV. [8 marks]