Before solving the following problems, make sure to install Codon and Seq by running these com- mands2:
bash -c "$(curl -fsSL https://exaloop.io/install.sh) "
export platform=$(uname -s | awk ' {print tolower($0)} ' )-$(uname -m)
cu![]() lta(-)r(L) z(h)x(t)v(t)f(p)s-:/-/C(g)i
lta(-)r(L) z(h)x(t)v(t)f(p)s-:/-/C(g)i![]()
![]() ucodon(b.com)/(/)lib(exa)
ucodon(b.com)/(/)lib(exa)![]() cod(oop)
cod(oop)![]() n(s)
n(s)![]() p(q)
p(q)![]() ugins(relea)ses/download/v0.11.3/seq-${platform} .tar.gz \
ugins(relea)ses/download/v0.11.3/seq-${platform} .tar.gz \
This time, you will need to use Python packages within Codon. Make sure to set up CODON_PYTHON environment variable to point to a working Python shared library. Use pip to install needed Python packages. If stuck, check this: https://xkcd.com/1987/. If that does not help, use Piazza to get help.
Then familiarize yourself with Codon (and Seq) by going through the following tutorials:
1. https://docs.exaloop.io/codon/general/intro
2. https://docs.seq-lang.org/tutorial/primer.html
3. https://docs.seq-lang.org/tutorial/tutorial.html
If you get stuck, let us know! We will be using Piazza to help you with Codon. Codon is still in development, so don’t be surprised if things do not work out! And please note down any bugs or
inconsistencies you discover during this assignment. General grievances and rants are also appreci- ated! You will submit all of those at the end of the semester as a part of Codon feedback collection program.
Each of these problems should be implemented as a short script in the Codon language unless otherwise specified. Each script should be relatively short (no more than 100 lines of code). The source code files and discussion points (in a PDF or a TXT, file please), should be submitted to Brightspace packed in a single ZIP file.
Problem 1 (Nopoly, 3 points): Implement a program that detects reads from a given FASTQ file whose canonical minimizer differs from their “Nopoly” minimizer. Print the read name, its canonical minimizer, and its Nopoly minimizer for each such read. If a read does not contain a Nopoly minimizer (e.g., read is a homopolymer), print - in its stead.
For this problem, assume that:
• a canonical minimizer is a minimal 14-mer within a read or its reverse complement; and
• a Nopoly minimizer is a minimal 14-mer (or its reverse complement) that contains no more than two identical nucleotides in a row (e.g. AAATATA is not a Nopoly minimizer because it contains AAA).
Example invocation:
# Get the data
$ ![]() ug(r)u(l)nz(-)ip(L)ft-c(p) :-/
ug(r)u(l)nz(-)ip(L)ft-c(p) :-/![]() ph(.)e(s)a(r)d(a) .e-n(b)4(i)0(.)a
ph(.)e(s)a(r)d(a) .e-n(b)4(i)0(.)a![]() .t(u)e(k)
.t(u)e(k)![]() t(v)ofq(l1)/fastq/SRR870/SRR870667/SRR870667_1.fastq.gz \
t(v)ofq(l1)/fastq/SRR870/SRR870667/SRR870667_1.fastq.gz \
$ codon run -plugin seq run nopoly.codon test.fq
|
SRR870667.1 |
AAACTACCAAGCAT |
AACTACCAAGCATT |
|
SRR870667.2 |
AAAAATAGGTTAAT |
AACGCAGTATTAAC |
|
SRR870667.3 |
AAAACAGATTGTAA |
AACAATCAATTACA |
|
SRR870667.4 |
AAAATGGTAGAGTG |
AACCTTCTGTTGAA |
|
SRR870667.5 |
AAAAAGGGCCTTTG |
AACTTCTTGATTGA |
|
SRR870667.6 |
AAAAATGGGATATG |
AAGGCAATTGGCAC |
|
SRR870667.7 |
AAACAGCCCCAGCA |
AACAGAGACTAACA |
|
SRR870667.8 |
AAAAAAATATCATT |
AACACCTCTAAGTT |
|
SRR870667.9 |
AAAAATGTACGAAA |
AAGCCTCATTATTC |
SRR870667.10 AAAAATCAGATCAT AACTCGGTAATAAT
Problem 2 (Euler, 4 points): Use the provided set of FASTA (not FASTQ!) reads assemble. fa that is (included with the assignment on Brightspace) to construct a de Bruijn graph (use any k-mer size for graph construction).
Assemble these reads by finding Eulerian walks in the constructed de Bruijn graph. Print the largest contig you have found. (Remember: each Eulerian walk in a graph corresponds to a contig.) Once you get the contig(s), use online BLAST to find out the source of the original sequence. Write the sequence source as a comment at the beginning of your source code.
Example invocation:
# Run the program
$ codon run -plugin seq run euler.codon reads.fa
ATTAAA . . .
# Make sure that the answer is in the source code
$ head euler.codon
## V012345678 euler
# The assembled contig is a genome of Some.species . . .
Note: Different k-mer sizes for de Bruijn graph construction might give you vastly different results. I got the best results with 12-mers; however, you are free to use other k-mer sizes. The implementation details of the Eulerian path might wildly impact the results, and you might even get different outputs across different runs. All of this is fine, so do not worry about it!
Problem 3 (Overlap, 3 points): Given a small FASTA file, generate and plot an overlap graph from its reads. Use Graphviz to generate overlap. png. Two nodes are connected if they have a perfect prefix-suffix match of size 10 or more (and the edge weight is the length of the maximal prefix-suffix match between the corresponding reads). Display the edge weight next to each edge.
Hint 1: You need to install Graphviz and the corresponding Python package (on Ubuntu/WSL, do apt install graphviz and pip3 install graphviz).
Hint 2: Here is an example Codon code that plots some nodes:
from python import graphviz
g = graphviz .Digraph(format= ' png ' )
for src, dst in edges:
g .edge(src, dst, label= ' weight ' )
g .render( ' overlap ' )
More Graphviz examples can be found here.
Hint 3: Use str type instead of seq to make your life easier.
Hint 4: Check https://docs.exaloop.io/codon/interoperability/pythonif stuck with Python support.
Example invocation:
# Plot the reads from "overlap.fa" (found at Brightspace)
$ codon run -plugin seq run overlap.codon overlap.fa
# Check if the file exists
$ ls overlap.png # should exist
Here is a sneak peek of the final image:
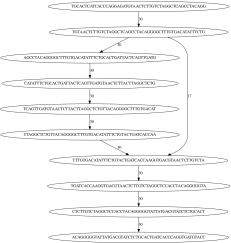
Bonus (2 points): Output all contigs by traversing the overlap graph.
Problem 4 (Allele, 4 points): Given a SAM file and a corresponding reference genome, analyze the alignments and output heterozygous and non-reference homozygous genotypes. A locus is heterozy- gous if it is covered with at least two alleles, each covering more than 40% of the reads at the locus. A locus is homozygous if an allele there has more than 80% of the total coverage.
Example invocation:
# Get the reference genome
$ wget -c http://hgdownload.soe.ucsc.edu/goldenPath/hg19/chromosomes/chr22.fa.gz
$ gunzip chr22.fa.gz
# Get the data
$ samtools view -h \
https://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase3/data/HG00275/alignment/\
HG00275.mapped.ILLUMINA.bwa.FIN.low_coverage.20120522.bam \
22:40000000-40002000 > test.sam
$ codon run -plugin seq run allele.codon chr22.fa test.sam
chr22 40000749 HET A/T
chr22 40000920 HOM T/T
chr22 40001316 HET G/T
chr22 40001320 HOM G/G
chr22 40001324 HET C/T
chr22 40001361 HET A/G
chr22 40001472 HOM G/G
chr22 40001533 HET C/G
chr22 40001541 HET A/T
chr22 40001544 HET C/T
chr22 40001997 HOM T/T
chr22 40002002 HOM G/G
Warning: A reference genome might contain lowercase letters. Make sure to cast them to uppercase letters (use str. upper to do that) for easier comparison.
Hint: Make sure to correctly parse SAM/BAM data by making use of CIGAR strings when ana- lyzing the alignments. Here is an example of how to do that:
1 ref, ref_start = ' ... ' , line .pos
2 read, read_start = ' ... ' , 0
3 for sz, op in line .cigar:
4 match op:
5 case ' M ' | ' = ' | ' X ' :
6 # matches/mismatches are between ref[ref_start:ref_start + sz] 7 # and read[read_start:read_start + sz]
8 # Indels and paddings are ignored;
9 # however, you still need to shift the reference/read positions
10 case ' I ' | ' S ' : read_pos += sz
11 case ' D ' | ' N ' : ref_pos += sz
12 case _ : pass
Problem 5 (Bloom, 3 points): Count all 30-mers in a given FASTA file! Use a counting Bloom filter with at least three different hash functions for efficient counting. Output all k-mers whose approximate count is greater than the second command-line argument.
Warning: Any attempt to use Dict[Kmer[30], int] to store or count k-mers will result in zero points. The same goes for other ingenious tricks (e.g. using list. count and so on). Use Bloom filters and do not store the whole set of k-mers anywhere, even temporarily! (You can store a constant number of k-mers, like those whose count exceeds the threshold.)
Hint: You can use MurmurHash family of hash functions. Here is a Codon implementation that you can use. Pick seeds as you please. Any reasonable output will be accepted.
1 def murmur (key, seed):
2 m, r = 0xc6a4a7935bd1e995 , 47
3 h = seed ˆ (8 * m)
5 k ˆ= k >> r; k *= m
6 h ˆ= k
7 h *= m; h ˆ= h >> r
8 h *= m; h ˆ= h >> r
9 return h
Example invocation:
# Get the data
$ samtools faidx \
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz \ 22:40000000-42000000 \
> test.fa
$ codon run -plugin seq bloom.codon test.fa 230
241 GGCTCACGCCTGTAATCCCAGCACTTTGGG
239 CTGTAATCCCAGCACTTTGGGAGGCCGAGG
238 GCTCACGCCTGTAATCCCAGCACTTTGGGA
235 CCTGTAATCCCAGCACTTTGGGAGGCCGAG
233 CAAAGTGCTGGGATTACAGGCGTGAGCCAC
231 CTCACGCCTGTAATCCCAGCACTTTGGGAG
Bonus (1 point): What would be the optimal choice of Bloom filter parameters? Why?
Bonus problems
Please submit the bonus problems via Brightspace.
Problem 6 (EMA, 10 points): EMA is a read aligner for 10X Genomics barcoded data. Read the EMA paper. Use it to align one million reads from some 10X dataset (some datasets are available here). How many barcodes did EMA find? How many did it correct? How much time it took you to get from FASTQs to BAMs? Is the alignment process easy or not? What could be improved?
Problem 7 (Assembly, 10 points): Get some small FASTQ dataset (E. coli or S. cerevisiae) from ENA or NCBI. Make sure to pick a species/strand that has a reference genome– you will need it! Try to run Velvet, SPAdes, and SGA assemblers on this dataset. Calculate N50 scores. Which assembler is the best? Why? How good were the assemblies in general? What did you learn about the assembly process?